一、临床表现
Wilson病患者可以在任何年龄起病,但多见于5~35岁[6],也有3岁起病的肝硬化患者[7]或80岁才出现症状的患者[8]。约有3%~4%的患者发病年龄晚于40岁[6, 9, 10]。
(一)神经精神表现
神经精神症状多见于10~30岁起病的患者[10, 11],主要表现为:(1)肌张力障碍;(2)震颤;(3)肢体僵硬和运动迟缓;(4)精神行为异常;(5)其他少见的神经症状。多个神经精神症状常同时出现,各个症状的轻重可能不同。神经精神症状的发生经常迟于肝脏症状,因此易被误诊为肝性脑病。
1.肌张力障碍:肌张力障碍早期可以是局灶、节段性的,逐渐发展为全身性,呈扭转痉挛状态,晚期常并发肢体严重挛缩。口面肌张力障碍较为常见,表现为构音困难、吞咽困难和流涎等。
2.震颤:多为姿势性或动作性震颤,最常见的是粗大不规则的震颤,也可见振幅较小的细颤,静止性搓丸样震颤较为少见。严重的姿势性震颤呈“扑翼样震颤”,易联想到肝性脑病、肺性脑病等代谢性脑病。
3.肢体僵硬和运动迟缓:部分患者可出现肢体僵硬、运动迟缓或减少、书写困难、写字过小、行走缓慢,易被误诊为帕金森病。
4.精神行为异常:精神行为异常在Wilson病患者中并不少见,甚至可早于肝脏损害和神经症状之前发生,却常被忽略。在青少年患者中,精神行为异常可表现为学习能力下降、人格改变、情绪波动、易激惹甚至性冲动等[12, 13]。在年长患者中,类偏执妄想、精神分裂症样表现、抑郁状态甚至自杀更为常见。尽管有研究报道Wilson病患者认知功能下降,但总体上不会发生明显的认知功能减退[14]。
5.其他少见的神经症状:少数患者可出现舞蹈样动作、手足徐动症、共济失调等神经症状。Wilson病患者发生癫痫并不罕见,可发生在疾病早期,更易发生在排铜治疗过程中[15, 16]。
(二)肝脏损害
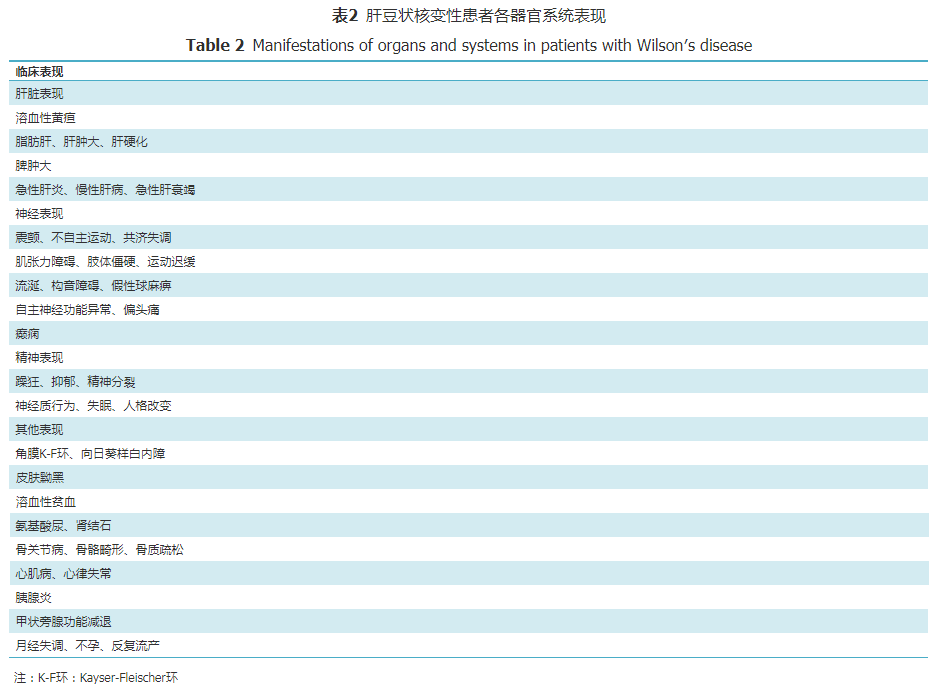
肝脏损害多见于婴幼儿及儿童患者,大部分患者在10~13岁起病[17],出现以下肝脏损害表现,如急性肝炎、暴发性肝衰竭、慢性肝病或肝硬化(代偿或失代偿)等(表2)[5]。
1.急性肝炎:患者可出现不明原因的黄疸、食欲差、恶心、乏力等急性肝炎症状,经护肝降酶等治疗可好转。
2.暴发性肝衰竭:少数患者可能突发急性肝衰竭即暴发性肝衰竭,其中部分患者伴有溶血性贫血,若不治疗,致死率高达95%[6]。即便经过排铜和护肝治疗,患者的肝功能仍可能急剧恶化。有文献报道,在因急性肝衰竭行急诊肝移植的患者中,Wilson病患者占6%~12%[18, 19]。
3.慢性肝病或肝硬化(代偿或失代偿):肝脏损害若未及时干预常常进展为慢性肝病或肝硬化。慢性肝病的临床症状缺乏特异性,常表现为黄疸、萎靡、腹胀、全身浮肿等。肝硬化可为代偿性或失代偿性,门脉高压性肝硬化亦可缺乏明显的临床症状而仅表现为脾肿大或血细胞减少。
(三)其他系统损害
铜离子蓄积在其他系统亦表现出相应的功能异常或损害,如肾脏损害[20]、骨关节病[21, 22]、心肌损害[23]、肌病[24]等。青年女性患者可出现月经失调、不孕和反复流产等。
(四)症状前个体
症状前个体一般指以下3种情况:常规体检发现转氨酶轻度增高但无症状且行ATP7B基因筛查确诊;意外发现角膜K-F环但无症状且行ATP7B基因筛查确诊;Wilson病先证者的无症状同胞行ATP7B基因筛查确诊。
二、诊断标准
主要是血清铜蓝蛋白降低,血清中非铜蓝蛋白的铜增多,尿铜排出量增加,肝含铜量增加。
1. 家族遗传史:父母是近亲婚配、同胞有HLD患者或死于原因不明的肝病者。
2. 缓慢进行性震颤、肌僵直、构语障碍等锥体外系症状、体征或/及肝症状。
3. 肉眼或裂隙灯证实有K-F环。
4. 血清铜蓝蛋白<200mg/L或铜氧化酶<0.2OD。
5. 尿铜>1.6μmol/24h。
6. 肝铜>250μg/g(干重)。
判断:
① 凡完全具备上述1--3项或2及4项者,可确诊为临床显性型。
② 仅具有上述3--5项或3--4项者属无症状型HLD。
③ 仅有1、2项或1、3项者,应怀疑HLD。
注:a:怀疑有神经系统受累时应行头颅MRI检查
三、治疗原则
WD治疗原则是尽早治疗、个体化治疗和终生治疗。